Sobre Cavernoma
Mal Formações Cavernosas e Epilepsias
DRA. SONIZA LEON, MD, PhD
Centro de Epilepsia dos Serviços de Neurologia e Neurocirurgia do Hospital Universitário Clementino Fraga Filho da
Universidade Federal do Rio de Janeiro
(CETEP-HUCFF/UFRJ)
A epilepsia é definida como uma desordem cerebral caracterizada pela predisposição de gerar crises epilépticas e pelas consequências neurobiológicas, cognitivas, psicológicas e sociais dessa condição. Acomete cerca de 0,8% da população mundial e é a mais comum entre as doenças neurológicas crônicas adquiridas, mas sua prevalência não é totalmente conhecida, variando em diferentes populações1. Depois da cefaléia, é a segunda condição crônica neurológica mais frequente em atendimentos de pacientes externos vistos por neurologistas.
A crise epiléptica é definida como “a ocorrência transitória de sinais e/ou sintomas devidos à atividade neuronal cerebral sincrônica ou excessiva” 3,6. O diagnóstico de epilepsia requer a ocorrência de pelo menos uma crise epiléptica3. É considerado síndrome epiléptica um conjunto complexo de sinais e sintomas que definem uma condição única e que pode envolver mais do que apenas o tipo de crise. Síndromes epilépticas têm sido identificadas por meio das características dos tipos de crises, do padrão de recorrência das crises, da idade de início, dos sinais neurológicos ou clínicos associados, dos achados eletroencefalográficos (EEG), pela existência ou ausência de ocorrência familiar e dos diferentes tipos de prognóstico.
As crises epilépticas são o sintoma mais frequente de pacientes com Malformações Cavernosas Cerebrais (CCMs) supratentoriais e podem evoluir para epilepsia fármaco-resistente em 40% dos casos. As malformações cavernosas (CCM) são lesões bem definidas, na maioria das vezes únicas, e estão presentes em 0.4 - 0.9% da população7. CCMs são malformações vasculares de aparência multilobulada que consistem de cavernas revestidas por tecido endotelial e essas dilatações de canais vasculares apresentam uma imagem característica na ressonância magnética (RM). Cerca de 48% das CCMs são diagnosticadas incidentalmente por RM realizadas por outros motivos, mas as crises epilépticas são a segunda causa mais comum de apresentação inicial (mais de 25% dos casos)8-10. O risco de crise recorrente após a primeira crise espontânea é de 94%, o que implica no fato de que o tratamento com fármacos antiepilépticos deve ser logo iniciado.
Por alterarem a anatomia e circuitos neurais locais e adjacentes, as crises epilépticas nas CCMs são as manifestações mais frequentes na evolução natural da doença7. Enquanto a maioria dos estudos sobre CCMs investiga o risco de hemorragias intracranianas, o controle das crises pode diminuir de forma significativa a incapacidade e melhorar a qualidade de vida desses pacientes, devendo ser um dos principais focos na condução do tratamento11.
Dentro do contexto das epilepsias, as CCMs estariam entre as causas de crises provocadas por uma doença de base conhecida. As epilepsias são denominadas de sintomáticas quando resultam de um ou mais distúrbios patológicos identificáveis na estrutura cerebral, ou no metabolismo, ou idiopática, presumidamente genética12, quando não existe um distúrbio conhecido5,6. Existem diferentes tipos de crises epilépticas e algumas podem representar uma entidade diagnóstica única ou serem classificadas com base definida por um substrato anatômico e fisiopatológico específico. No novo conceito e terminologia propostos pela Liga Internacional contra Epilepsia (InternationalLeagueAgainstEpilepsy – ILAE) a classificação das epilepsias focais, como presumidas de uma determinada região, deve ser evitada e é recomendado a descrição o mais detalhada possível da semiologia da crise.12 As CCMs estão entre as causas de epilepsias sintomáticas focais, mas estudos de séries também mostram que as CCMs podem ser assintomáticas 9.
Figura 1. Resumo da Revisão da terminologia para organização de crises e epilepsias da ILAE de 2010 divulgado durante o 29º Congresso da ILAE/LBE em agosto de 20116,12
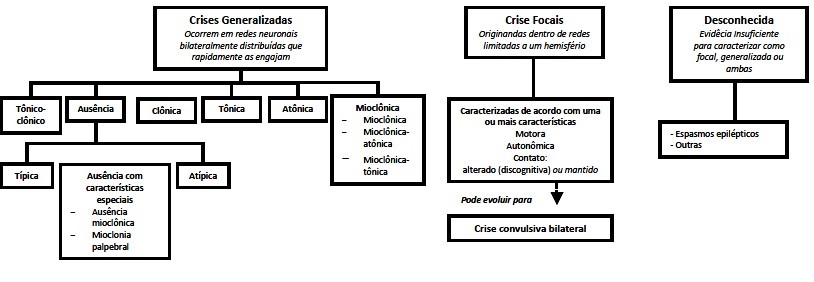
As crises epilépticas como manifestação das CCM estão entre as crises focais, originadas dentro de redes neurais limitadas (Figura 1). Como as CCMs podem ser múltiplas, diferentes redes neurais podem participar da manifestação epiléptica. Crises focais são aquelas originadas de algum ponto da circuitaria neuronal limitada a um hemisfério cerebral, e nenhuma classificação topográfica específica é recomendada. É recomendado, no entanto, que as crises sejam descritas com grande acurácia e de acordo com as suas características semiológicas, sem tentativa de enquadrá-las dentro de uma possível errônea categoria.12
A nova classificação proposta pela Liga Internacional contra Epilepsia (InternationalLeagueAgainstEpilepsy – ILAE) e publicada por Berg et al., 2010, contempla as diferentes etiologias das epilepsias focais, entre estas, aquelas com base nas alterações estruturais/metabólicas identificadas como fator que aumente o risco para o desenvolvimento de epilepsia, de origem genética ou adquirida. As CCMs estariam dentro deste grupo etiológico das epilepsias por serem uma doença geneticamente determinada e que secundariamente aumentam o risco de epilepsia.
FISIOPATOLOGIA DAS CRISES EPILÉPTICAS
O conhecimento atual mostra que as causas do fenômeno ictal (da crise epiléptica ou do foco epiléptico) não são únicas e estáticas, mas quase sempre evoluem ao longo do tempo13-15. Nas CCMs a base da epileptogênese parece estar associada à participação de fenômenos hipóxico-isquêmicos, neoangiogenesis e ação de mecanismos neuroinflamatórios com participação de metaloprotease 9 (MMP9) 21.
A base biológica das CCMs é dinâmica, com surgimento de novas lesões ao longo do tempo e hemorragias recorrentes. As características histológicas incluem a dilatação de canais vasculares, sem intervirem no parênquima cerebral, com paredes de vasos sanguíneos finas contendo endotélio, colágeno e adventícia, calcificações ou ossificações são vistas microscopicamente.
Na periferia, depósito de hemossiderina e macrófagos, estão quase sempre presentes o que contribui no diagnóstico diferencial com outras malformações vasculares. A presença de marcadores imunohistoquímicos de angiogenese e de proliferação como o fator de crescimento endotelial vascular (VEGF) e o antígeno nuclear celular de proliferação (PCNA)22 sugerem que as CCMs não são uma doença estática e que, sim, recorrem ao longo da vida em múltiplas regiões do sistema nervoso central, em diferentes momentos, como confirmam a clínica e os exames de RM longitudinais.
A relação entre lesões epileptogênicas e a extensão da epileptogencidade é um dos maiores desafios na avaliação pré-cirúrgica de pacientes com CCMs e epilepsia fármaco-resistentes. A investigação da zona epileptogênica por eletrodos profundos de estereoelectroencephalografia (SEEG) em pacientes com EFR associada a CCMs mostra que essa zona pode ser maior do que a própria lesão da CCM23. O efeito de massa das lesões das CCM parece não ter papel isolado na epileptogênese. A recorrência de microhemorragias e depósito de hemossiderina ao redor do tecido cortical parecem estar associados à hiperexcitabilidade neuronal devido à geração de radicais livres e peróxido de lipídio pelos íons de ferro24, é um dos principais indicativos da ocorrência de lesão estrutural25.
O mecanismo neuroinflamatório é outro fator, possivelmente, associado à epileptogênese13,26. A participação de MMP9, uma protease associada à vulnerabilidade da barreira hematoencefálica nas epilepsias do lobo temporal associada à esclerose hipocampal27,28 e encontrada nas CCMs, pode estar associada à migração transendotelial de moléculas do sangue periférico que teriam ação epileptogênica sobre os astrocitos que, sabidamente, apresentam papel nos mecanismos excitatórios e inibitórios das redes neurais29,30.
Casos isolados de Esclerose Hipocampal e CCMs são outro exemplo da co-exitência de fenômenos patológicos associados às epilepsias fármaco-resistentes 31. A astrogliose marcante encontrada nas CCMs é outro fator associado a epileptogênese; cada vez mais o papel dos astrocitos e sua interação na liberação de neurotransmissores excitatórios vem sendo associadas às epilepsias 14,26,32.
A recente inclusão da diferenciação entre malformações isoladas e combinadas foi uma das maiores novidades no consenso da ILAE de 2011 para as displasias corticais focais (FCD)33. Nas FCD tipo IIIc são encontradas anormalidades corticais laminares junto a lesões vasculares. A investigação por biópsia de 42 pacientes com EFR e CCMs encontrou 4.3% de FCD IIIce esta foi significativamente maior em pacientes com múltiplas CCMs33. Como as CCMs são lesões congênitas resultantes de distúrbio da diferenciação do mesoderma entre a terceira e oitava semana de gestação, a associação com anomalias venosas sugerem que compartilham dos mesmos mecanismos patogênicos precoces. CCMs e FCD são causas reconhecidas de EFR e casos anedóticos da associação entre essas condições vêm sendo relatados34.
Recente revisão sistemática de CCMs em pacientes com epilepsias submetidos a cirurgia investigou a presença concomitante de FCD; a ressecção da CCM foi seguida de controle das crises na sua grande maioria, e aqueles que não ficaram livres de crises apresentavam FCD adjacente a lesão35. O achado de múltiplas anormalidades associadas a malformação na mesma região de uma epilepsia fármaco-resistente pode ser sugestivo de possíveis mecanismos comuns permeando alterações do desenvolvimento33-35. Dentro do conhecimento dinâmico dos mecanismos associados à epileptogênese, a associação de mais de um fator de risco para o desenvolvimento de epilepsia pode contribuir em estratégias terapêuticas futuras.
TRATAMENTO DA EPILEPSIA ASSOCIADA AS MALFORMAÇÕES CAVERNOSAS CEREBRAIS
O tratamento da epilepsia associadas a CCMs deve considerar as características de apresentação das crises epilépticas, a resposta inicial aos fármaco-antiepilépticos, e ainda, buscar sua otimização. Pacientes que apresentam crises focais, sem generalização secundária, assim como em outras causas de epilepsias, parecem apresentar maior chance de controle.
Três grupos de pacientes podem ser distinguidos:
- aqueles que apresentaram uma crise única e que não foram tratados;
- aqueles com crises recorrentes que apresentam boa resposta com fármacos antiepilépticos, mantendo-se livres de crises; e
- aqueles que evoluem com epilepsia fármaco-resistente.
As séries de casos analisadas na literatura incluem quase exclusivamente a indicação e a resposta ao tratamento cirúrgico das epilepsies fármaco-resistentes de pacientes com CCMs.
Algorítmos de tratamento publicados incluem populações heterogêneas e não permitem conclusões claras. Mas estudo recente publicado por uma força tarefa da ILAE, especificamente, para epilepsias associadas a malformações cavernosas propôs uma definição diagnóstica clara e tratamentos com fármaco-antiepilépticos considerando a situação específica e as características individuais de cada paciente7. Os autores separaram pacientes definidos nas seguintes categorias, reproduzidos a partir dos estudos de risco de crises conduzido por Josephson et al., 201124:
1. Paciente com CCM descoberta de forma incidental ou CCM se apresentando com hemorragia intracraniana ou déficit neurológico focal – O risco de crises epilépticas em 5 anos após a descoberta incidental de CCM é de 4%, nos casos de CCM se apresentando com hemorragia intracraniana ou déficit neurológico focal é de 6%. A força tarefa da ILAE, baseada nesses números, considera que não é necessário iniciar tratamento profilático com fármacos-antiepilépticos em pacientes recém diagnosticados e que nunca apresentaram crises epilépticas24.
2. Pacientes com CCM que apresentam a doença com crise epiléptica única ou múltiplas.
2.1. Em pacientes que apresentam a primeira crise associada a CCM o risco de epilepsia nos 5 anos seguintes é de 94%, e o tratamento com fármacos antiepilépticos pode ser iniciado24. É recomendado atenção para outras manifestações recorrentes associadas a CCM, incluindo fenômenos não epilépticos ou crises psicogênicas, e o diagnóstico diferencial deve ser feito por meio de registro desses eventos.
De acordo com os guidelines da Cavernoma Alliance UK, 2012 e do National Institute for Health & Care Excellence, 2012, a força tarefa da ILAE reforça a recomendação para que todos os pacientes com CCM que apresentem uma crise epiléptica sejam, de forma urgente, encaminhados para um especialista com treinamento e experiência em epilepsia com o objetivo de investigar se as crises, ou as manifestações recorrentes apresentadas, estão relacionadas com a CCM.
Anamnese e registro histórico da semiologia das crises, história anterior de outros fenômenos possivelmente de natureza ictal, eletrencefalograma com as ativações de protocolo (registro em sono e vigília), e, na dependência da frequência desses fenômenos, registro video-eletrencefalográfico vão contribuir no diagnostico diferencial com outras causas de crises não-epilépticas como crises psicogênicas, síncope vaso-vagal, entre outros7,24.
Outro fator importante a ser considerado é a indicação de cirurgia nestes casos. A maioria dos autores é favorável a um tratamento conservador, com fármacos antiepilépticos no tratamento de crises recém diagnosticadas, ao invés de indicação de cirurgia 22. Não há estudo randomizado que permita afirmar se a cirurgia precoce versus tratamento conservador nas epilepsias associadas a CCM seria melhor, mas a história natural da doença mostra um curso em geral benigno, principalmente naqueles pacientes acompanhados clinicamente sem intervenção cirúrgica eletiva 22.
2.2. Pacientes com CCM recém diagnosticada que apresentaram crise epiléptica apresentam controle das crises com fármacos antiepilépticos entre 47-60% 24. Acompanhamento regular com neurologista é recomendado24.
2.3. Pacientes com CCM e crises epilépticas recorrentes – Esses pacientes entram dentro dos critérios de investigação pré-cirúrgica para epilepsias fármaco-resistentes, e, não precisariam passar por mais de um fármaco antiepiléptico para preencherem critérios de fármaco-resistência3,36. Pacientes com concordância entre paroxismos de pontas de projeção concordante com a área da CCM em estudo vídeo-eletrencefalográfico, e em área não eloquente também devem ser considerados como potenciais candidatos a cirurgia, mesmo que não apresentem crises durante a monitorização.
2.4. Paciente com CCM, predominantemente, em estruturas temporais associada a esclerose hipocampal e com concordância entre zona de início ictal (ZII) e as lesões morfo-estruturais, é recomendada a remoção da CCM associada ao amigadolo-hipocampectomia 31,37. Estudo recente corrobora a melhor resposta pós-cirúrgica no controle de crises em pacientes com esclerose hipocampal 31. Outro fator importante a ser considerado na janela de tempo terapêutico para intervenção cirúrgica é o fato de que nos pacientes com longos períodos de epilepsia refratária o controle de crises no pós-operatório é menor do que naqueles com crises menos frequentes (ILAE). O protocolo para indicação de cirurgia de epilepsia deve cumprir a investigação padrão para epilepsias fármaco-resistentes e nos casos onde a ZII não for concordante com as alterações da RM é recomendado a busca de eventuais lesões com exames de imagem de maior resolução, RM repetidas e outros como a tomografia por emissão de fóton único (SPECT). Em casos não concordantes com a ZII por vídeo-EEG de superfície, a investigação por eletrodos profundos é recomendada. Pacientes com descargas epileptiformes monofocais apresentam maior chance de controle das crises do que aqueles com descargas multifocais38,39.
2.5. Pacientes com CCM e patologia dual ou tripla 40 – Casos concomitantes de MFDC e de angiomas venosos têm sido relatados e a maioria desses pacientes não fica livre de crises se estas outras lesões morfoestruturais permanecerem. Rigoroso estudo pré-operatório e avaliação pré-cirúrgica cuidadosa podem minimizar o risco de um resultado não favorável no pós-operatório33,35,41.
2.6. A localização das CCMs parece estar relacionadas diretamente ao maior ou menor fator de risco para o surgimento de crises epilépticas. Pacientes com CCMs supratentoriais apresentam risco de crises entre 50-63% comparados aqueles com lesões infratentoriais entre 0-18%. CCMs corticais oferecem risco entre 57-70% quando comparadas com os 14-20% das lesões profundas supratentoriais7,24.
2.7. Em relação ao gênero, homens têm maior chance de controle das crises do que mulheres, provavelmente, pela exposição a fatores hormonais distintos.
2.8. O tratamento com fármacos antiepilépticos deve obedecer a indicação adotada para pacientes recém-diagnosticados com epilepsias focais com ou sem generalização secundária. Fármacos antiepilépticos de primeira linha são aqueles com pouca capacidade de auto indução enzimática, baixa ligação a proteínas plasmáticas e devem ser usados em monoterapia na posologia adequada para controle das crises. Entre estes, no Brasil, temos disponíveis a oxcarbazepina, a lamotrigina, o ácido varpróico, o divalproato de sódio, o topiramato, a gapapentina e a pregabalina. O levitiracetan e a lacosamida são fármacos antiepilépticos com eficácia nas epilepsias fármaco resistentes, e estão em fase de aprovação pela ANVISA, ainda não disponíveis no Brasil. A fenitoína, o fenobarbital e a carbamazepina também são fármacos que podem ser usados no tratamento das epilepsias focais com ou sem generalização secundária. O monitoramento laboratorial hematológico é ainda mais importante, pois alguns fármacos podem apresentar efeitos adversos que levam a maior risco de sangramento.
Podemos concluir que o manejo das epilepsias em pacientes com CCM requer cuidadosa análise das características individuais de cada caso baseado nas evidências conhecidas como fatores preditivos de maior ou de menor morbidade para o controle das crises epilépticas. As decisões na abordagem terapêutica, clínica ou de indicação cirúrgica, devem sempre ser tomadas por especialistas nesta área.
Referências Bibliográficas
1. Beghi E, Hesdorffer D. Prevalence of epilepsy-An unknown quantity. Epilepsia 2014; 55(7): 963-7.
2. Noronha AL, Borges MA, Marques LH, et al. Prevalence and pattern of epilepsy treatment in different socioeconomic classes in Brazil. Epilepsia 2007; 48(5): 880-5.
3. Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005; 46(4): 470-2.
4. Yamakawa K. Na channel gene mutations in epilepsy--the functional consequences. Epilepsy research 2006; 70 Suppl 1: S218-22.
5. Engel J, Jr., editor. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven Publishers; 1997.
6. Engel J, Jr. Report of the ILAE classification core group. Epilepsia 2006; 47(9): 1558-68.
7. Rosenow F, Alonso-Vanegas MA, Baumgartner C, et al. Cavernoma-related epilepsy: review and recommendations for management--report of the Surgical Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2013; 54(12): 2025-35.
8. Dorsch NWC, McMahon JHA. Intracranial cavernous malformations - natural history and management. Critical reviews in neurosurgery : CR 1998; 8(3): 154-68.
9. Gross BA, Lin N, Du R, Day AL. The natural history of intracranial cavernous malformations. Neurosurgical focus 2011; 30(6): E24.
10. Aiba T, Tanaka R, Koike T, Kameyama S, Takeda N, Komata T. Natural history of intracranial cavernous malformations. Journal of neurosurgery 1995; 83(1): 56-9.
11. Bascarevic V, Vojvodic N, Ristic AJ, et al. Epilepsy surgery outcome in temporal lobe cavernoma and multiple sclerosis. Clinical neurology and neurosurgery 2013; 115(10): 2178-81.
12. Berg AT, Scheffer IE. New concepts in classification of the epilepsies: entering the 21st century. Epilepsia 2011; 52(6): 1058-62.
13. Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nature reviews Neurology 2011; 7(1): 31-40.
14. Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiology of disease 2008; 29(1): 142-60.
15. Kassahun Y, Perrone R, De Momi E, et al. Automatic classification of epilepsy types using ontology-based and genetics-based machine learning. Artificial intelligence in medicine 2014; 61(2): 79-88.
16. Ito M, Nagafuji H, Okazawa H, et al. Autosomal dominant epilepsy with febrile seizures plus with missense mutations of the (Na+)-channel alpha 1 subunit gene, SCN1A. Epilepsy research 2002; 48(1-2): 15-23.
17. Yamakawa K. [Na channel gene mutations and epilepsy mouse model]. No to hattatsu Brain and development 2007; 39(3): 171-3.
18. Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nature genetics 1998; 19(4): 366-70.
19. Hedera P, Ma S, Blair MA, et al. Identification of a novel locus for febrile seizures and epilepsy on chromosome 21q22. Epilepsia 2006; 47(10): 1622-8.
20. Aronica E, Boer K, Redeker S, et al. Differential expression patterns of chloride transporters, Na+-K+-2Cl--cotransporter and K+-Cl--cotransporter, in epilepsy-associated malformations of cortical development. Neuroscience 2007; 145(1): 185-96.
21. Alvarez de Eulate-Beramendi S, Alvarez-Vega MA, Antuna-Ramos A, et al. [Pathogenetic bases of epileptogenesis in cerebral cavernomas]. Revista de neurologia 2012; 55(12): 718-24.
22. Awad I, Jabbour P. Cerebral cavernous malformations and epilepsy. Neurosurgical focus 2006; 21(1): e7.
23. Sevy A, Gavaret M, Trebuchon A, et al. Beyond the lesion: the epileptogenic networks around cavernous angiomas. Epilepsy research 2014; 108(4): 701-8.
24. Josephson CB, Leach JP, Duncan R, et al. Seizure risk from cavernous or arteriovenous malformations: prospective population-based study. Neurology 2011; 76(18): 1548-54.
25. de Souza JM, Domingues RC, Cruz LC, Jr., Domingues FS, Iasbeck T, Gasparetto EL. Susceptibility-weighted imaging for the evaluation of patients with familial cerebral cavernous malformations: a comparison with t2-weighted fast spin-echo and gradient-echo sequences. AJNR American journal of neuroradiology 2008; 29(1): 154-8.
26. Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nature reviews Neuroscience 2014; 15(1): 43-53.
27. Quirico-Santos T, Meira ID, Gomes AC, et al. Resection of the epileptogenic lesion abolishes seizures and reduces inflammatory cytokines of patients with temporal lobe epilepsy. Journal of neuroimmunology 2013; 254(1-2): 125-30.
28. Quirico-Santos T, Nascimento Mello A, Casimiro Gomes A, de Carvalho LP, de Souza JM, Alves-Leon S. Increased metalloprotease activity in the epileptogenic lesion--Lobectomy reduces metalloprotease activity and urokinase-type uPAR circulating levels. Brain research 2013; 1538: 172-81.
29. Diniz LP, Almeida JC, Tortelli V, et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor beta signaling through modulation of D-serine levels in cerebral cortex neurons. The Journal of biological chemistry 2012; 287(49): 41432-45.
30. Diniz LP, Tortelli V, Garcia MN, et al. Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia 2014.
31. Menzler K, Thiel P, Hermsen A, et al. The role of underlying structural cause for epilepsy classification: clinical features and prognosis in mesial temporal lobe epilepsy caused by hippocampal sclerosis versus cavernoma. Epilepsia 2011; 52(4): 707-11.
32. Foresti ML, Arisi GM, Shapiro LA. Role of glia in epilepsy-associated neuropathology, neuroinflammation and neurogenesis. Brain research reviews 2011; 66(1-2): 115-22.
33. Niehusmann P, Becker AJ, Malter MP, Raabe A, Bostrom A, von der Brelie C. Focal cortical dysplasia type IIIc associates with multiple cerebral cavernomas. Epilepsy research 2013; 107(1-2): 190-4.
34. Giulioni M, Zucchelli M, Riguzzi P, Marucci G, Tassinari CA, Calbucci F. Co-existence of cavernoma and cortical dysplasia in temporal lobe epilepsy. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 2007; 14(11): 1122-4.
35. Chen DJ, Severson E, Prayson RA. Cavernous angiomas in chronic epilepsy associated with focal cortical dysplasia. Clinical neuropathology 2013; 32(1): 31-6.
36. von der Brelie C, Schramm J. Cerebral cavernous malformations and intractable epilepsy: the limited usefulness of current literature. Acta neurochirurgica 2011; 153(2): 249-59.
37. Brelie C, von Lehe M, Raabe A, et al. Surgical resection can be successful in a large fraction of patients with drug-resistant epilepsy associated with multiple cerebral cavernous malformations. Neurosurgery 2014; 74(2): 147-53; discussion 53.
38. von der Brelie C, Kuczaty S, von Lehe M. Surgical management and long-term outcome of pediatric patients with different subtypes of epilepsy associated with cerebral cavernous malformations. Journal of neurosurgery Pediatrics 2014; 13(6): 699-705.
39. von der Brelie C, Malter MP, Niehusmann P, Elger CE, von Lehe M, Schramm J. Surgical management and long-term seizure outcome after epilepsy surgery for different types of epilepsy associated with cerebral cavernous malformations. Epilepsia 2013; 54(9): 1699-706.
40. Maciunas JA, Syed TU, Cohen ML, Werz MA, Maciunas RJ, Koubeissi MZ. Triple pathology in epilepsy: coexistence of cavernous angiomas and cortical dysplasias with other lesions. Epilepsy research 2010; 91(1): 106-10.
41. Johnson AM, Sugo E, Barreto D, et al. Clinicopathological associations in temporal lobe epilepsy patients utilising the current ILAE focal cortical dysplasia classification. Epilepsy research 2014.